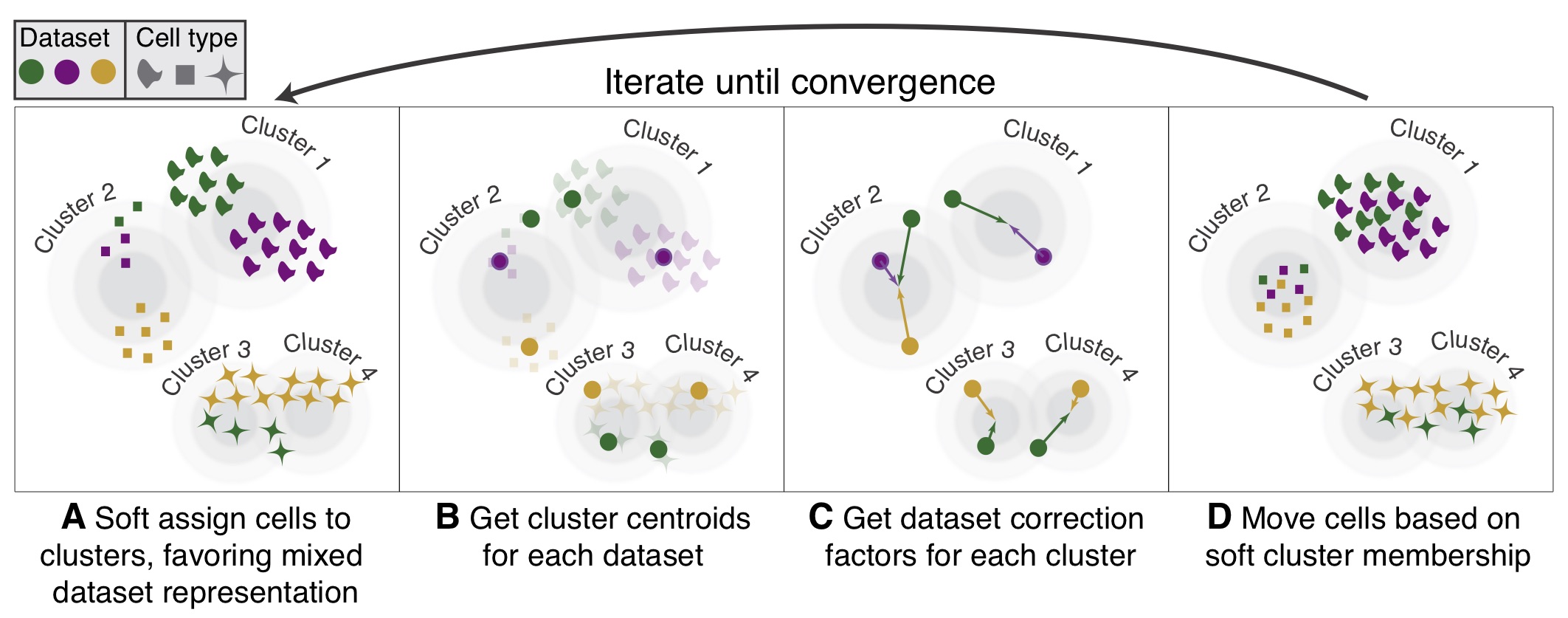
Harmony is a general-purpose R package with an efficient algorithm for integrating multiple data sets. It is especially useful for large single-cell datasets such as single-cell RNA-seq.
Harmony is:
- Fast: Analyze thousands of cells on your laptop.
- Sensitive: Different cell types may be present or absent in each batch.
- Accurate: Integrate cells from multiple donors, tissues – even different technologies.
Getting started
See how to use Harmony with your data and integrate it into your analysis pipeline.
Advanced tutorial
Find out more about the internal data structures and algorithm details in this tutorial.

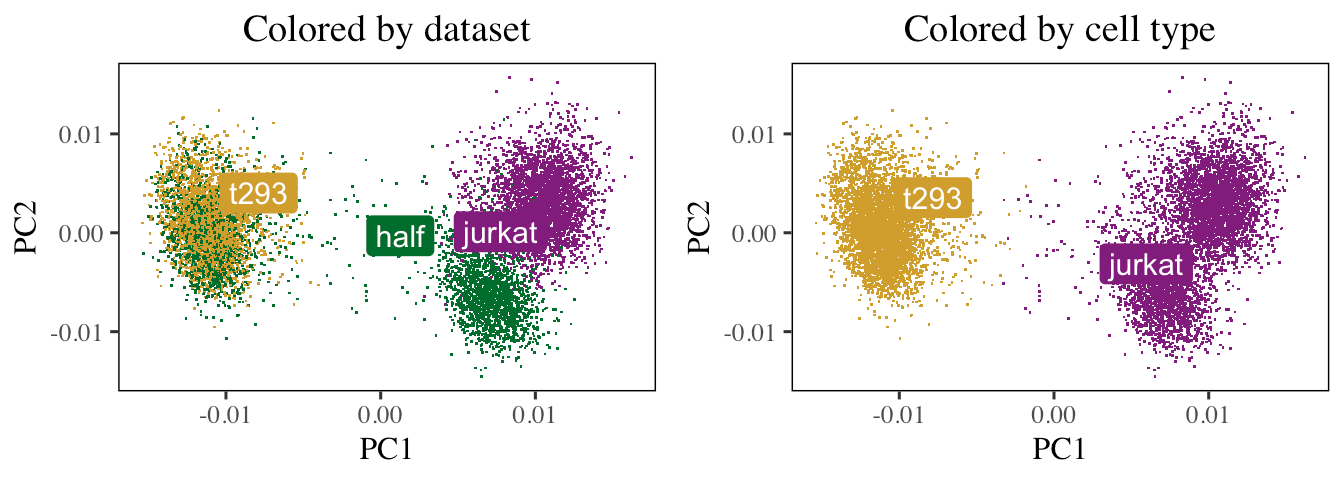
Animation
Visualize how Harmony aligns single-cell RNA-seq datasets from three different donors.
Installation
The easiest way to get Harmony is to install it from Github:
Harmony has been tested on R versions >= 3.4 on Linux, macOS, and Windows.
Usage
Run the HarmonyMatrix() function on your PCs from principal component analysis:
library(harmony)
harmonized_pcs <- HarmonyMatrix(
data_mat = pcs, # Matrix with coordinates for each cell (row) along many PCs (columns)
meta_data = meta_data, # Dataframe with information for each cell (row)
vars_use = "dataset", # Column in meta_data that defines dataset for each cell
do_pca = FALSE # Since we are providing PCs, do not run PCA
)Citation
If you use Harmony for published work, please cite our manuscript:
Fast, sensitive, and accurate integration of single cell data with Harmony
Ilya Korsunsky, Jean Fan, Kamil Slowikowski, Fan Zhang, Kevin Wei, Yuriy Baglaenko, Michael Brenner, Po-Ru Loh, Soumya Raychaudhuri
bioRxiv 2019. doi.org/10.1101/461954
We will share the code needed to reproduce results from the manuscript at https://github.com/immunogenomics/harmony2019.
Links
- Browse source code at
https://github.com/immunogenomics/harmony - Report a bug at
https://github.com/immunogenomics/harmony/issues
Citation
Developers
-
Ilya Korsunsky
Maintainer, author
- Nghia Millard
Author -
Soumya Raychaudhuri
Author - All authors...