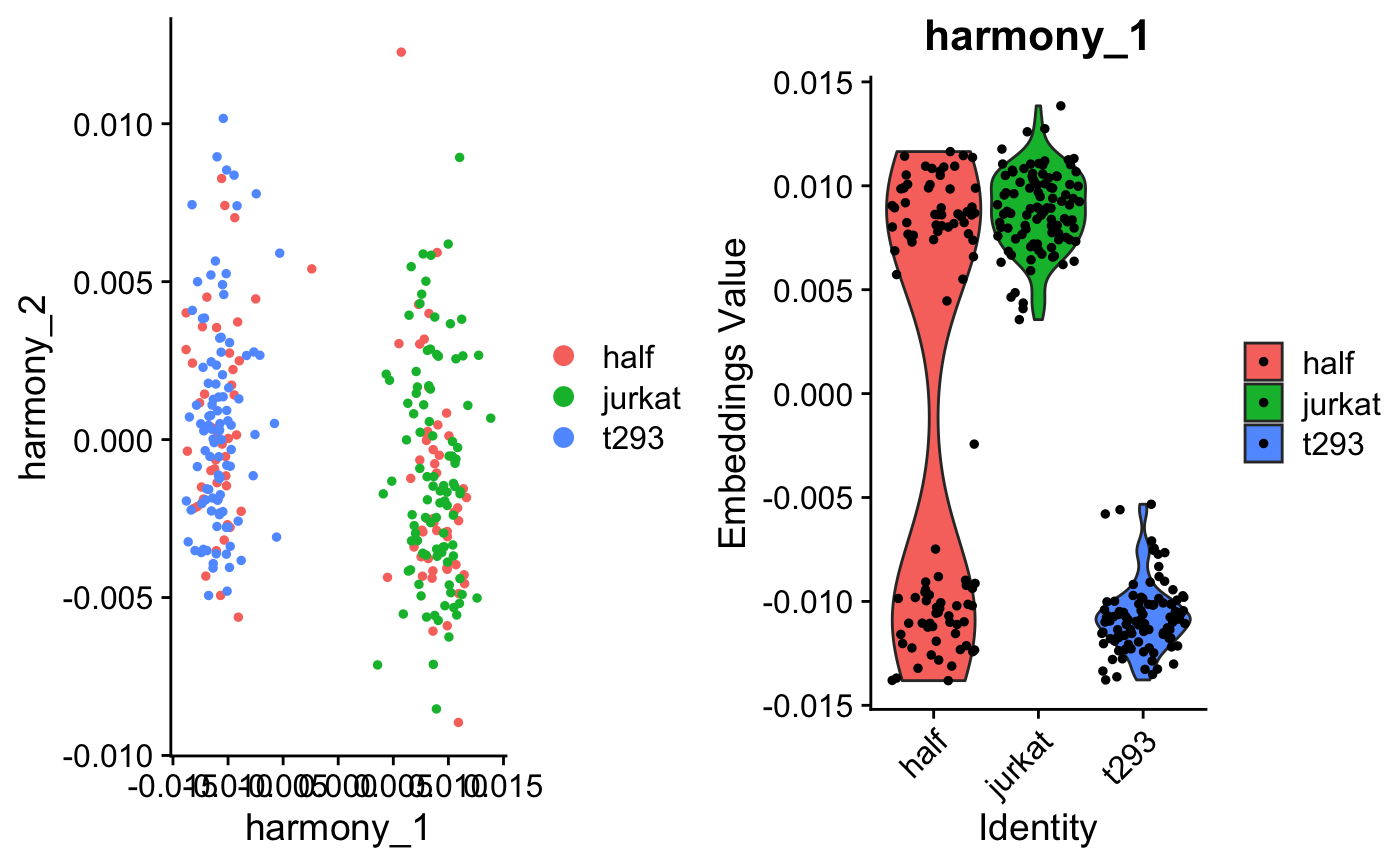
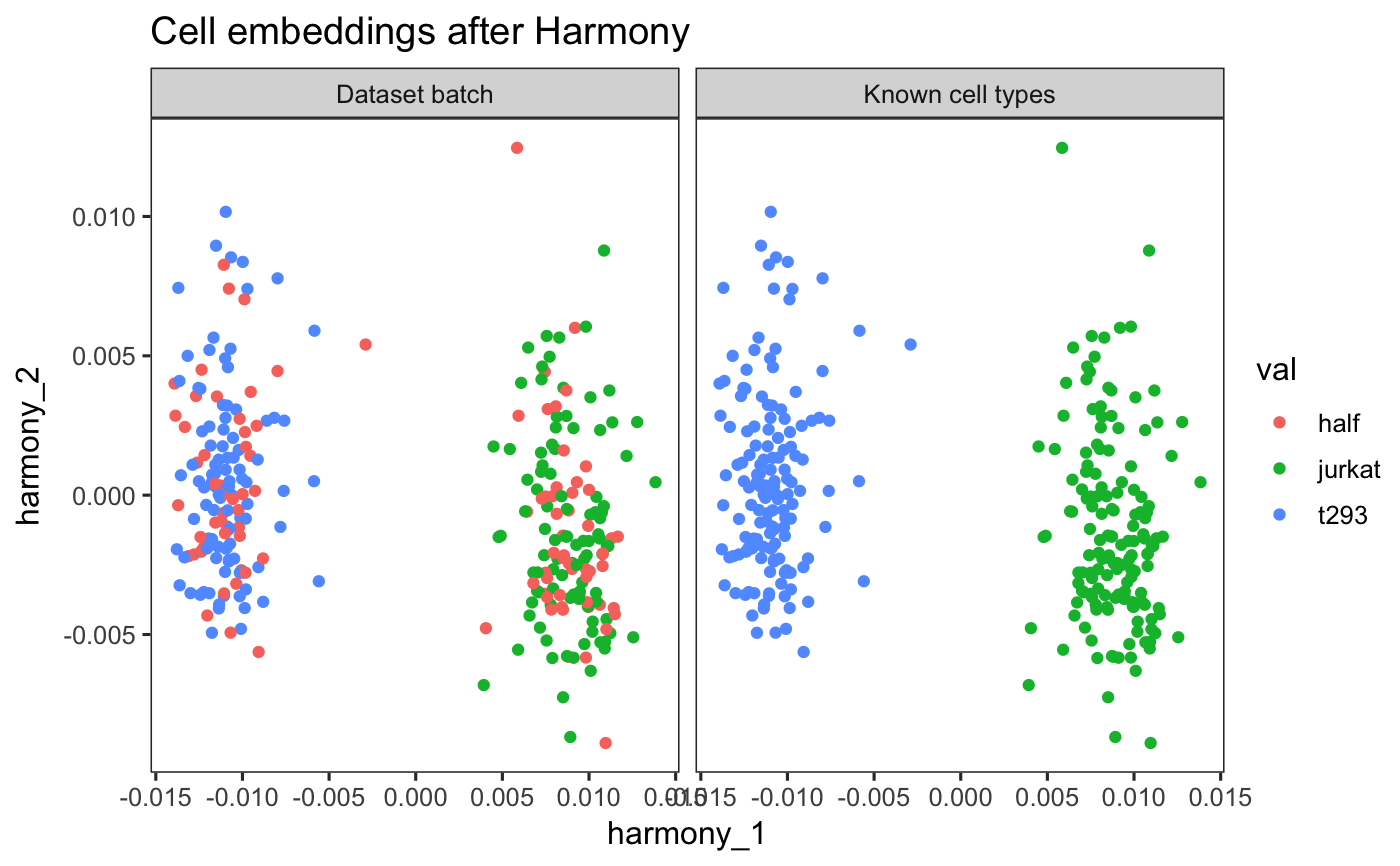
Run Harmony algorithm with Seurat and SingleCellAnalysis pipelines.
RunHarmony(object, group.by.vars, ...) # S3 method for seurat RunHarmony(object, group.by.vars, dims.use = NULL, theta = NULL, lambda = NULL, sigma = 0.1, nclust = NULL, tau = 0, block.size = 0.05, max.iter.harmony = 10, max.iter.cluster = 20, epsilon.cluster = 1e-05, epsilon.harmony = 1e-04, plot_convergence = FALSE, verbose = TRUE, reference_values = NULL, reduction.save = "harmony", ...) # S3 method for Seurat RunHarmony(object, group.by.vars, reduction = "pca", dims.use = NULL, theta = NULL, lambda = NULL, sigma = 0.1, nclust = NULL, tau = 0, block.size = 0.05, max.iter.harmony = 10, max.iter.cluster = 20, epsilon.cluster = 1e-05, epsilon.harmony = 1e-04, plot_convergence = FALSE, verbose = TRUE, reference_values = NULL, reduction.save = "harmony", assay.use = "RNA", project.dim = TRUE, ...) # S3 method for SingleCellExperiment RunHarmony(object, group.by.vars, dims.use = NULL, theta = NULL, lambda = NULL, sigma = 0.1, nclust = NULL, tau = 0, block.size = 0.05, max.iter.harmony = 10, max.iter.cluster = 20, epsilon.cluster = 1e-05, epsilon.harmony = 1e-04, plot_convergence = FALSE, verbose = TRUE, reference_values = NULL, reduction.save = "HARMONY", ...)
Arguments
- object
Pipeline object. Must have PCA computed.
- group.by.vars
Which variable(s) to remove (character vector).
- ...
other parameters
- dims.use
Which PCA dimensions to use for Harmony. By default, use all
- theta
Diversity clustering penalty parameter. Specify for each variable in group.by.vars. Default theta=2. theta=0 does not encourage any diversity. Larger values of theta result in more diverse clusters.
- lambda
Ridge regression penalty parameter. Specify for each variable in group.by.vars. Default lambda=1. Lambda must be strictly positive. Smaller values result in more aggressive correction.
- sigma
Width of soft kmeans clusters. Default sigma=0.1. Sigma scales the distance from a cell to cluster centroids. Larger values of sigma result in cells assigned to more clusters. Smaller values of sigma make soft kmeans cluster approach hard clustering.
- nclust
Number of clusters in model. nclust=1 equivalent to simple linear regression.
- tau
Protection against overclustering small datasets with large ones. tau is the expected number of cells per cluster.
- block.size
What proportion of cells to update during clustering. Between 0 to 1, default 0.05. Larger values may be faster but less accurate
- max.iter.harmony
Maximum number of rounds to run Harmony. One round of Harmony involves one clustering and one correction step.
- max.iter.cluster
Maximum number of rounds to run clustering at each round of Harmony.
- epsilon.cluster
Convergence tolerance for clustering round of Harmony Set to -Inf to never stop early.
- epsilon.harmony
Convergence tolerance for Harmony. Set to -Inf to never stop early.
- plot_convergence
Whether to print the convergence plot of the clustering objective function. TRUE to plot, FALSE to suppress. This can be useful for debugging.
- verbose
Whether to print progress messages. TRUE to print, FALSE to suppress.
- reference_values
(Advanced Usage) Defines reference dataset(s). Cells that have batch variables values matching reference_values will not be moved
- reduction.save
Keyword to save Harmony reduction. Useful if you want to try Harmony with multiple parameters and save them as e.g. 'harmony_theta0', 'harmony_theta1', 'harmony_theta2'
- reduction
Name of dimension reduction to use. Default is PCA.
- assay.use
(Seurat V3 only) Which assay to Harmonize with (RNA by default).
- project.dim
Project dimension reduction loadings. Default TRUE.
Value
Seurat (version 2) object. Harmony dimensions placed into dimensional reduction object harmony. For downstream Seurat analyses, use reduction.use='harmony' and reduction.type='harmony'.
Seurat (version 3) object. Harmony dimensions placed into dimensional reduction object harmony. For downstream Seurat analyses, use reduction='harmony'.
SingleCellExperiment object. After running RunHarmony, the corrected cell embeddings can be accessed with reducedDim(object, "Harmony").
Examples
## Seurat Version 2 if (requireNamespace("Seurat", quietly = TRUE)) { pkg_version <- packageVersion('Seurat') if (pkg_version >= "2.0" & pkg_version < "3.0") { data(cell_lines_small_seurat_v2) seuratObject <- RunHarmony(cell_lines_small_seurat_v2, 'dataset', lambda = .1, verbose = FALSE) ## Harmony cell embeddings harmony_embedding <- Seurat::GetCellEmbeddings( seuratObject, 'harmony' ) harmony_embedding[seq_len(5), seq_len(5)] ## Harmony gene loadings harmony_loadings <- Seurat::GetGeneLoadings( seuratObject, 'harmony' ) harmony_loadings[seq_len(5), seq_len(5)] p1 <- Seurat::DimPlot(seuratObject, reduction.use = 'harmony', group.by = 'dataset', do.return = TRUE) p2 <- Seurat::VlnPlot(seuratObject, features.plot = 'Harmony1', group.by = 'dataset', do.return = TRUE) cowplot::plot_grid(p1,p2) } } ## Seurat Version 3 if (requireNamespace("Seurat", quietly = TRUE)) { pkg_version <- packageVersion('Seurat') if (pkg_version >= "3.0" & pkg_version < "4.0") { data(cell_lines_small_seurat_v3) seuratObject <- RunHarmony(cell_lines_small_seurat_v3, 'dataset', lambda = .1, verbose = FALSE) ## Harmony cell embeddings harmony_embedding <- Seurat::Embeddings(seuratObject, 'harmony') harmony_embedding[seq_len(5), seq_len(5)] ## Harmony gene loadings harmony_loadings <- Seurat::Loadings(seuratObject, 'harmony') harmony_loadings[seq_len(5), seq_len(5)] p1 <- Seurat::DimPlot(seuratObject, reduction = 'harmony', group.by = 'dataset', do.return = TRUE) p2 <- Seurat::VlnPlot(seuratObject, features = 'harmony_1', group.by = 'dataset', do.return = TRUE) cowplot::plot_grid(p1, p2) } }#>## SingleCellExperiment if (requireNamespace("SingleCellExperiment", quietly = TRUE)) { data(cell_lines_small_sce) sceObject <- RunHarmony(cell_lines_small_sce, 'dataset', lambda = .1, verbose = FALSE) ## Harmony cell embeddings harmony_embedding <- SingleCellExperiment::reducedDim( sceObject, 'HARMONY' ) harmony_embedding[seq_len(5), seq_len(5)] ## Plot the Harmonized embeddings ## Colored by batch and cell type SingleCellExperiment::reducedDim(sceObject, 'HARMONY') %>% cbind(SingleCellExperiment::colData(sceObject)) %>% data.frame() %>% tidyr::gather(key, val, dataset, cell_type) %>% dplyr::mutate(key = dplyr::case_when( key == 'dataset' ~ 'Dataset batch', key == 'cell_type' ~ 'Known cell types' )) %>% dplyr::sample_frac(1L, FALSE) %>% ggplot2::ggplot(ggplot2::aes(x = harmony_1, y = harmony_2, color = val)) + ggplot2::geom_point() + ggplot2::facet_wrap(~key) + ggplot2::theme_test(base_size = 12) + ggplot2::labs(title = 'Cell embeddings after Harmony') }